
Enfermedad de Gaucher, una patología donde el "médico de AP presta una atención integral, accesible y longitudinal"
La enfermedad de Gaucher es una enfermedad rara que afecta a 1 de cada 100.000 personas, en el conjunto de la población general. En España hay alrededor de 500 pacientes diagnosticados y se producen, aproximadamente, 20 casos nuevos cada año. El 26 de julio se celebra el Día Mundial de la Enfermedad de Gaucher, en el que se ofrece apoyo a todos los pacientes y familias que conviven con esta patología y se insiste en la importancia de promover la investigación, para prever diagnósticos y tratamientos erróneos.
"Se trata de una enfermedad hereditaria, con un patrón de herencia autosómica recesiva. Esto quiere decir que, para que se padezca la enfermedad, ambos progenitores deben ser portadores o enfermos. Por ello, es más frecuente en poblaciones donde se registran más relaciones de consanguinidad", asegura el Dr. Sergio Capilla, Secretario del Grupo de Trabajo de Medicina Genómica Personalizada y Enfermedades Raras de SEMERGEN. La Enfermedad de Gaucher se engloba dentro del grupo de las llamadas enfermedades lisosomales. Los lisosomas son los orgánulos que tienen las células para hacer la digestión y ésta tiene por finalidad transformar las sustancias que tomamos en materias primas, para la construcción de nuevas estructuras celulares. En el interior de los lisosomas se encuentran diferentes enzimas encargadas de dicho proceso. Cuando una de ellas falla, bien porque no se produzca o bien porque funcione mal, desencadena, por una parte, un acúmulo de la sustancia a digerir y por otra, un déficit del producto de la digestión. En el caso de la enfermedad de Gaucher la enzima implicada se denomina beta-glucocerebrosidasa y su afectación produce un acúmulo de glucosilceramida en el interior de las células y tejidos, responsable de las manifestaciones clínicas de la enfermedad.
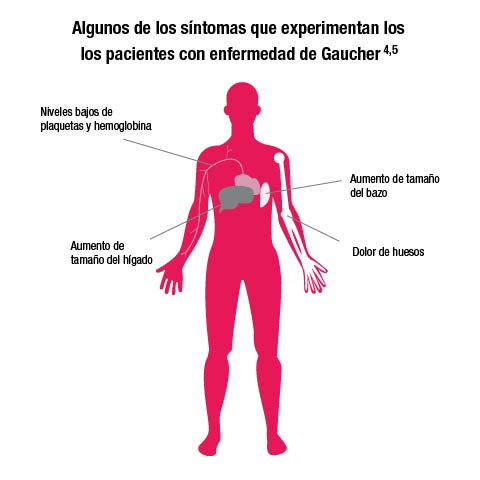
"La enfermedad de Gaucher afecta a todos los órganos y sus manifestaciones están relacionadas con la intensidad con que se produce el acúmulo de glucosilceramida, en cada uno de ellos", subraya el Dr. Capilla. Son frecuentes el aumento de tamaño del hígado y del bazo, lo que da lugar a dolores y molestias abdominales; la afectación de la médula ósea que origina anemia y hemorragia; del sistema nervioso que ocasiona alteraciones cognitivas, de la coordinación y del movimiento; del corazón, con afectación de las válvulas cardíacas y del sistema osteoarticular, siendo típicas las crisis de dolor óseo en la infancia, la osteoporosis prematura y el retraso del crecimiento. Clínicamente se diferencian tres tipos: el tipo I es la forma más frecuente y no afecta al cerebro. En el tipo II el sistema nervioso se ve afectado de manera más intensa y rápida, además de ser la forma más rara y severa de la enfermedad, al tiempo que conlleva un mal pronóstico en los primeros años de vida. En el tipo III la afectación neurológica es menos intensa y, al igual que en el tipo I, los pacientes pueden llegar sin diagnóstico a la edad adulta.
"Hasta final del siglo pasado, a estos pacientes sólo se les podía ofrecer tratamiento paliativo y de soporte. Sin embargo, en la actualidad disponemos de tratamientos efectivos que reemplazan o modifican la enzima deficitaria y logran frenar la progresión de la enfermedad, permitiendo al paciente tener una vida normal".
El médico de Atención Primaria presta una atención integral (centrada en la persona como tal), coordinada con otros profesionales, accesible y longitudinal (a lo largo de la vida). Por ello, el papel del médico de familia es especialmente relevante y podemos resumirlo en cinco áreas:
- Evitar el retraso en el diagnóstico y terapéutico: Son muchos pacientes los que presentan una sintomatología inespecífica e insidiosa que condiciona un retraso en el diagnóstico de muchos años, y, por ende, del tratamiento específico.
- Manejo y seguimiento de la enfermedad y de sus complicaciones.
- Acompañamiento del paciente y su familia desde un abordaje psicosocial. La información, la escucha y la empatía son necesidades que deben ser cubiertas por el médico de familia.
- Asesoramiento reproductivo.
- Registro epidemiológico. Por una parte, proporcionar información al Sistema Sanitario de la dimensión del problema permite su reorientación hacia las necesidades reales y por otra, facilita al paciente su participación.
El Grupo de Trabajo de Medicina Genómica, Personalizada y Enfermedades Raras de SEMERGEN, sin menoscabo de la actividad investigadora, mantiene una actitud proactiva en la información y formación del médico de familia en estas áreas. "Por último, es sumamente importante que el paciente y su familia conozcan la importancia del movimiento asociativo que, a nivel nacional, puede estar representado por la Asociación Española de Enfermos y Familiares de la Enfermedad de Gaucher (AEEFEG) o por la Federación Española de Enfermedades Raras (FEDER)".
